Las encefalopatías espongiformes (enfermedades priónicas) son aquellas enfermedades en las que las formas patológicas de las proteínas priónicas están involucradas en el desarrollo. Sabemos cada vez más sobre las enfermedades priónicas, pero los aspectos clave aún se desconocen; actualmente, la medicina no tiene los medios para curar a los pacientes de estas enfermedades.
Las encefalopatías espongiformes, o enfermedades priónicas, se pueden desarrollar durante la vida, mientras que otras surgen de mutaciones genéticas heredadas presentes desde el nacimiento. Dentro de este grupo, hay varias entidades que ocurren en humanos, ejemplos son la enfermedad de Creutzfeldt-Jakob o el insomnio familiar fatal.
Las enfermedades priónicas han sido muy misteriosas durante mucho tiempo. A diferencia de otros patógenos, como bacterias, virus u hongos, no contienen ácido nucleico: los priones están hechos solo de proteínas. La teoría de las enfermedades priónicas fue descubierta por S. Prusiner, este descubrimiento fue muy apreciado en la comunidad científica; en 1997, el investigador recibió el Premio Nobel de Medicina. Aunque han pasado relativamente muchos años desde que nació el concepto de priones, algunos científicos todavía creen que está incompleto y están investigando más la naturaleza de estas afecciones; algunos de los factores responsables de las encefalopatías espongiformes ahora se han confirmado.
Enfermedades por priones: causas
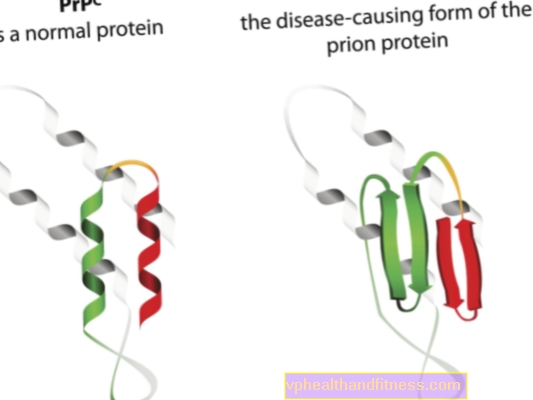
La etiología de las enfermedades priónicas está relacionada con la transformación de proteínas priónicas normales en formas patógenas y patógenas. Los priones son moléculas de proteínas que se encuentran en el cuerpo de todo ser humano. Su función aún no está del todo clara, pero se sabe que, en condiciones normales, las proteínas priónicas no dañan el organismo. La situación es diferente cuando los priones cambian su estructura y se convierten en partículas patógenas, entonces se desarrolla una de varias encefalopatías espongiformes. Los priones que se encuentran naturalmente en el cuerpo se denominan PRPC, mientras que las formas anormales se denominan PRPSC. Estos últimos son un problema grave no solo porque pueden acumularse en el tejido nervioso en forma de depósitos y generar daños en él, sino también porque tienen la capacidad de transformar priones normales en una forma malformada (en pocas palabras, PRPSC puede "infectar" proteínas normales con su potencial patógeno).
Lea también: Enfermedad de Huntington (corea de Huntington): causas, síntomas, tratamiento Temblores musculares - causas. ¿Qué significa temblor muscular? Enfermedades que matan más rápido: CHOQUE, ÉBOLA, MALDICIÓN, ATAQUE, EMERGENCIA [GALE ...Básicamente, existen 3 causas de encefalopatías espongiformes:
- esporádico (la mutación patógena ocurre en las células somáticas, ocurre durante la vida del paciente),
- familia (resultante de la carga de mutaciones heredadas de los padres),
- Paso (relacionado con la introducción de priones patógenos en el cuerpo humano, por ejemplo, a través de preparaciones de hormona del crecimiento contaminadas con estas partículas o trasplante de córnea de una persona que padece alguna encefalopatía espongiforme).
Encefalopatías espongiformes: enfermedad de Creutzfeldt-Jakob
La enfermedad de Creutzfeldt-Jakob (CJD) se describió por primera vez a principios de la década de 1920. Hay 4 tipos de enfermedades:
- ECJ esporádica (la más común, representa hasta 9/10 de todos los casos de ECJ)
- ciudad natal de CJD
- abrumado por CJD
- variante de CJD
El cuadro clínico en el curso de diversas variantes de la enfermedad de Creutzfeldt-Jakob puede ser variable. Las dolencias más frecuentes en el curso de este grupo de encefalopatías espongiformes son:
- trastornos de demencia (incluido el deterioro progresivo de la memoria, la atención y la concentración)
- mioclonías (movimientos involuntarios como sacudidas repentinas de los músculos)
- disfunción cerebelosa (manifestada, por ejemplo, por trastornos del equilibrio)
- visión borrosa
- síntomas piramidales y extrapiramidales
En el curso de las variantes de CJD, también pueden aparecer trastornos mentales (por ejemplo, ansiedad, estado de ánimo deprimido), dolor y otros movimientos involuntarios distintos de los mencionados anteriormente.
El pronóstico de la enfermedad de Creutzfeldt-Jakob es malo; por ejemplo, en pacientes con ECJ esporádica, se tarda un promedio de cuatro a cinco meses desde el inicio de los síntomas de la enfermedad hasta la muerte.
Encefalopatías espongiformes: síndrome de Gerstmann-Straussler-Scheinker
El síndrome de Gerstmann-Straussler-Scheinker (GSS) suele ser hereditario y es causado por una mutación hereditaria en el gen PRNP. Se considera que es la encefalopatía espongiforme de progresión más lenta. El equipo de GSS incluye:
- ataxia espinocerebelosa
- disartria
- trastornos de demencia
- trastornos de la deglución
- nistagmo
- aumento de la tensión muscular
Los pacientes diagnosticados con GSS tienen un período de tiempo variable y, en algunos pacientes, la muerte ocurre más de 10 años después del inicio.
Encefalopatías espongiformes: insomnio familiar fatal
El insomnio familiar letal es una enfermedad priónica causada por mutaciones en el gen PRNP. La enfermedad es extremadamente rara y hasta ahora se ha diagnosticado en 28 familias en todo el mundo. En el curso del insomnio familiar fatal, el primer síntoma es la incapacidad para dormir. Este problema da como resultado trastornos de ansiedad y el paciente experimenta alucinaciones. El efecto de la falta constante de descanso nocturno son alteraciones en el funcionamiento del sistema autónomo (incluidos cambios en la función cardíaca, sudoración y trastornos del sistema digestivo), también hay una disminución progresiva del peso corporal. En etapas más avanzadas del insomnio familiar fatal, aparecen alteraciones hormonales y aparecen síntomas de demencia en el curso de la enfermedad.
El pronóstico para el insomnio familiar fatal, al igual que para otras encefalopatías espongiformes, es malo: los pacientes generalmente mueren dentro de los tres años posteriores al inicio.
Encefalopatías espongiformes: prionopatía con susceptibilidad variable a la proteasa
La aparición de estas encefalopatías espongiformes se relaciona principalmente con mutaciones en el gen PRNP. Sin embargo, estas mutaciones se refieren a diferentes codones de este gen y, por lo tanto, se distinguen varias enfermedades priónicas diferentes. Una unidad descrita relativamente recientemente (en 2008) es la prionopatía con susceptibilidad variable a la proteasa. Las personas que padecen esta enfermedad portan mutaciones en hasta tres codones del gen PRNP.
En la prionopatía con susceptibilidad variable a la proteasa, los pacientes experimentan:
- deterioro cognitivo
- severidad extrema de los trastornos psiquiátricos: pueden ser euforia y agitación, pero también apatía significativa
- disartria
- afasia (trastornos del lenguaje)
La duración media de la enfermedad en esta prionopatía es inferior a 4 años.
Encefalopatías espongiformes: kuru
Kuru ahora se considera una enfermedad que prácticamente ya no existe: se encontró en representantes de tribus de Papúa Nueva Guinea, que practicaban un comportamiento caníbal. El síntoma dominante de esta encefalopatía espongiforme es la ataxia cerebelosa progresiva. Puede ir acompañada de movimientos involuntarios (principalmente en forma de corea, temblores y atetosis), así como incontinencia urinaria y fecal. Los pacientes que toman kuru también experimentan cambios de humor significativos, desarrollan reflejos primitivos (por ejemplo, chupar). Un problema bastante característico en el caso de esta enfermedad priónica son los episodios forzados de llanto o risa; debido a este último fenómeno, a veces se hace referencia al kuru como "muerte por risa".
Encefalopatías espongiformes: diagnóstico
Las enfermedades priónicas pueden sospecharse sobre la base de los síntomas del paciente. Sin embargo, son bastante inespecíficos, ya que también pueden aparecer en el curso de una serie de otras enfermedades que no están relacionadas con los priones. Por esta razón, los siguientes también se utilizan en el diagnóstico de encefalopatías espongiformes:
- pruebas de imagen (por ejemplo, resonancia magnética, que permite detectar cambios relacionados con la degeneración del cerebro por las proteínas priónicas),
- pruebas de laboratorio (como la evaluación de las concentraciones de proteínas en el líquido cefalorraquídeo, por ejemplo, MAP-tau, S-100 o proteínas 14-3-3),
- pruebas genéticas (para detectar la presencia de mutaciones en el paciente),
- Pruebas inmunhistoquímicas (que utilizan anticuerpos contra las proteínas priónicas).
El diagnóstico también puede confirmarse mediante autopsia del cerebro, en la que es posible encontrar cambios característicos de las encefalopatías espongiformes. Pueden ser lesiones esponjosas, de distribución diversa y con una estructura diferente (dependiendo de una entidad patológica específica), placas amiloides y defectos neuronales.
Encefalopatías espongiformes: tratamiento
Actualmente, las enfermedades priónicas son incurables; a pesar de los numerosos estudios que se han realizado durante muchos años, la medicina aún no cuenta con fármacos que puedan ralentizar o inhibir completamente su progreso. El tratamiento sintomático se utiliza en pacientes con encefalopatías espongiformes, cuyo objetivo es aliviar la intensidad de los síntomas y mejorar al máximo su calidad de vida.
Sin embargo, aún se está trabajando en el tratamiento de las encefalopatías espongiformes. Los científicos están tratando de utilizar varios métodos; el primer ejemplo es la terapia génica. Afectarían a los ácidos nucleicos y las mutaciones presentes en su estructura; el propósito de aplicar la terapia génica sería neutralizar errores en el código genético. Un enfoque diferente es la base de la terapia inmunológica: se está trabajando para crear anticuerpos cuya función sería eliminar los priones patógenos. Otro método que ve el potencial para combatir las encefalopatías espongiformes es el tratamiento con el uso de moléculas de proteínas sintetizadas que, una vez introducidas en el cuerpo del paciente, neutralizarían las proteínas patológicas.
Articulo recomendado:
Encefalopatías: causas, tipos y síntomas






-przepisy-tworzce-jadospis-w-diecie-dr-kwaniewskiego.jpg)