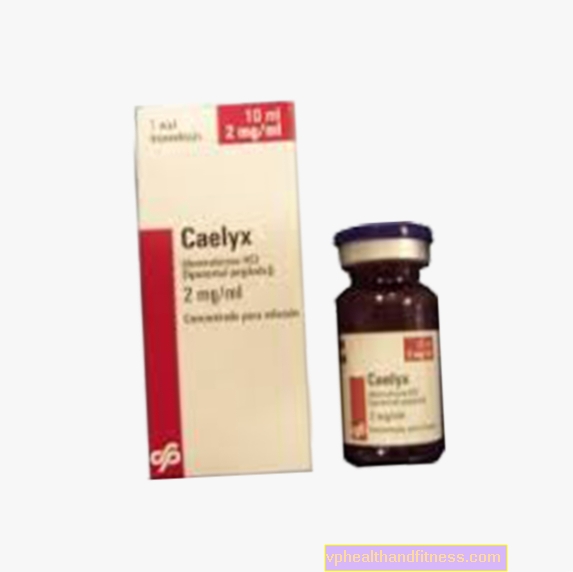
1 ml de la preparación contiene 2 mg de clorhidrato de doxorrubicina en liposomas pegilados. La preparación contiene sacarosa y fosfatidilcolina de soja completamente hidrogenada (de soja).
| Nombre | Contenido del paquete | La sustancia activa | Precio 100% | Última modificación |
| Caelyx | 1 vial, final por preparar solución a inf. | Clorhidrato de doxorrubicina | 2019-04-05 |
Acción
Antibiótico citotóxico antraciclina obtenido de cultivos de Streptomyces peucetius var. cesio. El fármaco se acumula entre pares de bases adyacentes en la doble hélice del ADN, lo que evita que se despliegue necesario para la replicación. Esto conduce a una inhibición de la síntesis de ADN, ARN y proteínas. La forma liposomal pegilada de hidrocloruro de doxorrubicina prolonga el tiempo de residencia del fármaco en el sistema circulatorio. La farmacocinética de la preparación difiere significativamente de las formas estándar de doxorrubicina. A dosis más bajas (10 mg / m2 - 20 mg / m2), la preparación muestra una farmacocinética lineal; en el rango de dosis de 10 mg / m2. - 60 mg / m2 la farmacocinética no es lineal. La preparación, a diferencia de las formas estándar, que se distribuyen en gran medida en los tejidos, permanece principalmente en el volumen del líquido vascular y la eliminación de la doxorrubicina de la sangre depende del portador liposomal. La doxorrubicina está disponible después de que los liposomas abandonan el vaso y entran en el compartimento de tejido. Después de la administración de dosis equivalentes de la preparación y formas estándar, la concentración en sangre y los valores de AUC de la forma liposomal pegilada son mayores que los obtenidos con las formas estándar de clorhidrato de doxorrubicina. T0,5 es de 24-231 h, con una media de 73,9 h.
Dosis
Por vía intravenosa, por infusión. Administrar solo bajo la supervisión de un oncólogo especialista con experiencia en el uso de fármacos citotóxicos. La preparación no se puede usar indistintamente con otras formas farmacéuticas de clorhidrato de doxorrubicina. Cáncer de mama o de ovario: 50 mg / m2 cada 4 semanas mientras la enfermedad no progrese y la paciente tolera el tratamiento. Mieloma múltiple: 30 mg / m2 en el día 4 de un ciclo de tratamiento de bortezomib de 3 semanas como una infusión de 1 hora inmediatamente después de la infusión de bortezomib. El régimen de tratamiento con bortezomib es de 1,3 mg / m2 los días 1, 4, 8 y 11 durante ciclos de tratamiento de 3 semanas. El tratamiento debe continuar mientras se mantenga la respuesta al tratamiento mientras el paciente lo tolere. El día del tratamiento combinado (día 4 del ciclo) puede posponerse a 48 horas si está médicamente indicado, pero el intervalo entre dosis consecutivas de bortezomib no debe ser inferior a 72 horas SIDA Sarcoma de Kaposi: 20 mg / m2. Cada 2-3 semanas Deben evitarse interrupciones inferiores a 10 días, ya que no se puede descartar la acumulación de fármaco y una mayor toxicidad. Se recomienda que el tratamiento se continúe durante 2-3 meses. El tratamiento debe continuarse según sea necesario para mantener una respuesta terapéutica. Modificación de la dosis en caso de reacciones adversas. Para controlar los efectos secundarios (como enrojecimiento palmar y de las plantas de los pies - EPI, estomatitis o toxicidad hematológica), la dosis puede reducirse o administrarse más tarde. Eritrodisestesia palmoplantar (PPE). 1er. 4 semanas después de la dosis anterior de la preparación: se debe administrar el 100% de la dosis si el paciente no experimentó ninguna tercera o cuarta toxicidad cutánea previa, y si ocurrió, espere una semana más. 1er. toxicidad en la quinta semana después de la dosis anterior de la preparación: se debe administrar el 100% de la dosis si el paciente no experimentó ninguna tercera o cuarta toxicidad cutánea previa, y si ocurrió, espere una semana más el primer día. toxicidad en la sexta semana después de la dosis anterior de la preparación: reduzca la dosis en un 25%; volver al descanso de 4 semanas. 2º. Toxicidad (eritema, descamación o hinchazón que interfiere con, pero no previene, la actividad física normal; pequeñas ampollas o úlceras con un diámetro de estomatitis. Primera toxicidad (ulceración indolora, eritema o dolor leve) en la cuarta semana después de la dosis anterior de la preparación - debe administrar el 100% de la dosis si el paciente no desarrolló estomatitis antes de la 3ra o 4ta estomatitis, y si el paciente no tuvo una estomatitis previa, espere una semana adicional 1 de toxicidad en la 5ta semana después de la dosis anterior de la preparación - se debe administrar el 100% de la dosis, si no la estomatitis anterior ocurrió en la tercera o cuarta semana, y si ocurrió - espere una semana adicional de la primera semana de toxicidad en la sexta semana después de la dosis anterior de la preparación - reduzca la dosis en un 25%; vuelva a la pausa de 4 semanas o, según la evaluación del médico, suspenda la administración 2.a toxicidad (eritema doloroso, hinchazón o ulceración, pero con posibilidad de comer) en la 4.a semana después de la dosis anterior del preparado - espere una semana más z. 2st. toxicidad en la quinta semana después de la dosis anterior de la preparación - espere una segunda semana adicional. toxicidad en la sexta semana después de la dosis anterior de la preparación: reduzca la dosis en un 25%; vuelva al descanso de 4 semanas o suspenda la dosificación según el criterio de su médico. 3º. Toxicidad (eritema doloroso, hinchazón o ulceración sin posibilidad de comer) en la cuarta semana después de la dosis anterior de la preparación: espere una semana más. toxicidad en la quinta semana después de la dosis anterior de la preparación: espere una semana más. 6 semanas después de la dosis anterior de la preparación: suspenda la administración. 4º. toxicidad (nutrición parenteral o enteral requerida) en la cuarta semana después de la dosis anterior de la preparación; espere una semana adicional. toxicidad en la quinta semana después de la dosis anterior de la preparación - espere una cuarta semana adicional. 6 semanas después de la dosis anterior de la preparación: suspenda la administración. El esquema de modificación de dosis anterior también es aplicable a pacientes con sarcoma de Kaposi de SIDA y pacientes con mieloma múltiple que reciben terapia combinada con bortezomib. Efecto tóxico sobre el sistema hematopoyético (cáncer de mama o de ovario) - 1º: RAN (número absoluto de neutrófilos) 1500-1900 / mm3, plaquetas 75.000-150.000 / mm3 - reanudar el tratamiento sin reducción de dosis; 2º: ANC 1000 - 3, plaquetas 50.000 - 3, esperar hasta que ANC ≥ 1.500 / mm3 y plaquetas ≥ 75.000 / mm3, volver a administrar sin reducción de dosis; 3ª etapa: RAN 500 - 3, plaquetas 25.000 - 3 - esperar hasta RAN ≥ 1.500 / mm3 y plaquetas ≥ 75.000 / mm3, volver a administrar sin reducción de dosis; 4º: RAN 3, plaquetas 3 - espere hasta que RAN ≥ 1.500 / mm3 y plaquetas ≥ 75.000 / mm3, reduzca la dosis en un 25% o continúe el tratamiento con la dosis completa con factor de crecimiento. Efecto tóxico sobre el sistema hematopoyético (pacientes con sarcoma de Kaposi en el curso del sida): el tratamiento con la preparación debe interrumpirse temporalmente cuando el ANC es 3 y / o el recuento de plaquetas es 3, simultáneamente, para aumentar el recuento de glóbulos cuando el ANC es 3, en ciclos posteriores. Se puede administrar G-CSF (o GM-CSF). Modificación de la dosis para pacientes con mieloma múltiple en tratamiento combinado con bortezomib. Fiebre ≥38 ° C y ANC 3: no administre la dosis adecuada de doxorrubicina si los síntomas aparecieron antes del cuarto día del ciclo de tratamiento; si después del día 4, la siguiente dosis debe reducirse en un 25%; Reduzca la siguiente dosis de bortezomib en un 25%. En cualquier día de administración después del primer día de cada ciclo: recuento de plaquetas 3: no administre la dosis requerida de doxorrubicina si los síntomas aparecieron antes del cuarto día del ciclo de tratamiento; para los síntomas después del día 4, la dosis debe reducirse en un 25% en los ciclos posteriores si la dosis de bortezomib se reduce debido a toxicidad hematológica; no administre la dosis correcta de bortezomib. Si se retienen 2 o más dosis de bortezomib en un ciclo de tratamiento para ciclos posteriores, reduzca la dosis en un 25%. Desarrollo de toxicidad no hematológica de grado 3 o 4: no administrar una dosis de doxorrubicina hasta que la afección mejore a grado de niños y adolescentes. La experiencia en niños es limitada. Por esta razón, no se recomienda su uso en pacientes menores de 18 años. Grupos especiales de pacientes. Pacientes con insuficiencia hepática. Inicio del tratamiento: si el nivel de bilirrubina se encuentra entre 1,2 y 3,0 mg / dL, la primera dosis debe reducirse en un 25%. Si la bilirrubina es> 3,0 mg / dL, la primera dosis debe reducirse en un 50%. Si el paciente tolera la primera dosis sin aumentar la bilirrubina o las enzimas hepáticas, la dosis en el segundo ciclo puede aumentarse al siguiente nivel de dosis, es decir, si la primera dosis se ha reducido en un 25%, entonces la dosis debe aumentarse a la dosis completa en el segundo ciclo; si la primera dosis se reduce en un 50%, la dosis debe aumentarse al 75% de su valor total en el segundo ciclo. La dosis puede aumentarse a su valor total en ciclos posteriores. En pacientes con metástasis hepáticas asociadas con un aumento de la bilirrubina y las enzimas hepáticas, el fármaco puede usarse hasta 4 veces el límite superior de lo normal. No es necesario ajustar la dosis en pacientes de edad avanzada y en pacientes con insuficiencia renal; no se dispone de datos farmacocinéticos en pacientes con CCr. Forma de administración. No debe administrarse por vía intramuscular o subcutánea. No administrar en bolo o solución sin diluir. Se recomienda que el equipo de infusión se conecte mediante una rama lateral del catéter a la infusión intravenosa de solución de glucosa al 5% (50 mg / ml) para obtener una mayor dilución y minimizar el riesgo de trombosis y extravasación. La infusión se puede administrar en una vena periférica. No utilice filtros de infusión en línea. Para dosis <90 mg: diluir la preparación en 250 ml de solución de glucosa al 5% (50 mg / ml) para perfusión. Para dosis ≥90 mg: diluir la preparación en 500 ml de solución de glucosa al 5% (50 mg / ml) para perfusión. Para el cáncer de mama / cáncer de ovario / mieloma múltiple, la primera dosis debe administrarse a una velocidad de no más de 1 mg / minuto para minimizar el riesgo de reacciones a la infusión. Si no hay ninguna reacción relacionada con la perfusión, se pueden administrar más perfusiones en 60 minutos. Los pacientes que experimentan una reacción a la perfusión deben modificar la vía de perfusión de la siguiente manera: administrar el 5% de la dosis total mediante perfusión lenta durante los primeros 15 minutos. Si la perfusión se tolera sin respuesta, la velocidad de administración puede duplicarse durante los próximos 15 minutos. Si la perfusión aún se tolera, la perfusión se puede finalizar dentro de otra hora por un tiempo total de perfusión de 90 minutos. En el caso del sarcoma de Kaposi en el curso del sida, la dosis de la preparación se diluye en 250 ml de solución para perfusión de glucosa al 5% (50 mg / ml) y se administra como perfusión intravenosa durante 30 minutos.
Indicaciones
Monoterapia del cáncer de mama metastásico en pacientes con mayor riesgo de complicaciones cardíacas. Tratamiento del cáncer de ovario avanzado en pacientes cuya quimioterapia de primera línea con compuestos de platino ha fracasado. Tratamiento de pacientes con progresión de mieloma múltiple en terapia combinada con bortezomib que hayan recibido al menos una línea de tratamiento anterior y que ya se hayan sometido o no sean elegibles para un trasplante de médula ósea. Tratamiento del sarcoma de Kaposi (SK) relacionado con el SIDA en pacientes con recuentos bajos de CD4 (menos de 200 / mm3) con afectación significativa de las membranas mucosas, la piel o los órganos internos. La preparación se puede utilizar en pacientes con SK-SIDA en quimioterapia de primera o segunda línea, cuando se ha observado progresión de la enfermedad a pesar de la terapia de combinación utilizada anteriormente que consiste en al menos dos de los siguientes medicamentos: alcaloides de la vinca, bleomicina y la forma farmacéutica estándar de doxorrubicina (u otra antraciclina) o sin tolerancia.
Contraindicaciones
Hipersensibilidad al principio activo, cacahuetes o soja. No debe utilizarse en pacientes con sarcoma de Kaposi de SIDA para quienes el tratamiento local o sistémico con interferón alfa puede ser eficaz.
Precauciones
Debido a las diferencias en los perfiles farmacocinéticos y los esquemas de dosificación, la preparación no debe usarse de manera intercambiable con otros medicamentos que contengan clorhidrato de doxorrubicina. Se recomiendan pruebas de ECG de rutina frecuentes mientras toma el medicamento. Se debe considerar la biopsia de miocardio si se produce una reducción del complejo QRS. De forma rutinaria antes de iniciar el tratamiento con la preparación y repetida periódicamente durante el tratamiento, se recomienda la medición ecocardiográfica de la fracción de eyección del ventrículo izquierdo o la angiografía de múltiples cuadros (MUGA). La evaluación de la función ventricular izquierda se considera obligatoria antes de cada administración de fármaco adicional que supere una dosis acumulada de 450 mg de antraciclinas / m2. durante la vida. Durante el tratamiento con antraciclina, el mencionadoLas pruebas y métodos de evaluación del rendimiento cardíaco deben usarse en el siguiente orden: registro de ECG, medición de la fracción de eyección del ventrículo izquierdo, biopsia endomiocárdica. Debido al efecto cardiotóxico del fármaco, se debe tener especial cuidado en pacientes con enfermedades cardíacas, incluida la insuficiencia cardíaca, y en pacientes que reciben otras antraciclinas. La dosis total de doxorrubicina HCl debe tener en cuenta cualquier tratamiento previo (o simultáneo) con agentes cardiotóxicos (incluidos: otras antraciclinas, antraquinonas o, por ejemplo, 5-fluorouracilo); un grupo de riesgo adicional son los pacientes que previamente han sido sometidos a irradiación mediastínica o que reciben ciclofosfamida concomitante, cuya cardiotoxicidad también puede ocurrir después de una dosis acumulada de antraciclinas inferior a 450 mg / m2. El perfil de seguridad cardíaca de la pauta posológica recomendada para el tratamiento del cáncer de mama y de ovario (50 mg / m2) es similar al de la dosis de 20 mg / m2. en pacientes con sarcoma de Kaposi relacionado con el SIDA. Debido a la posibilidad de trastornos de la médula ósea, se deben realizar hemogramas frecuentes durante el tratamiento (antes de cada dosis). La disfunción grave persistente de la médula ósea puede provocar superinfecciones y sangrado. Se han observado leucemias mieloides agudas secundarias y mielodisplasias en pacientes que reciben terapia combinada con doxorrubicina; Todo paciente que reciba doxorrubicina debe estar bajo control hematológico. Debido a los casos de cáncer oral secundario, tanto durante el tratamiento como hasta 6 años después de la última dosis, los pacientes deben ser controlados regularmente para detectar úlceras o molestias en la boca. Debido a la posibilidad de reacciones alérgicas y anafilactoides graves y a veces potencialmente mortales poco después de comenzar la perfusión (con síntomas como: asma, rubor, urticaria, dolor de pecho, fiebre, hipertensión, taquicardia, picazón, sudoración, dificultad para respirar, hinchazón). escalofríos, dolor de espalda, opresión en el pecho y la garganta y / o hipotensión, convulsiones), la primera dosis debe administrarse a una velocidad de no más de 1 mg / min. Cada vial de la preparación contiene sacarosa y el medicamento se administra en una solución de glucosa al 5%, lo que debe considerarse en pacientes con diabetes. Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis y es esencialmente "exento de sodio".
Actividad indeseable
Los efectos secundarios más comúnmente observados en el cáncer de mama o de ovario son eritrodisestesia palmoplantar - EPP (los casos totales fueron 44-46,1%; el tratamiento se interrumpió en algunas pacientes por EPP grave) y estomatitis o mucositis y náuseas. . En pacientes con sarcoma de Kaposi relacionado con el SIDA, se ha observado con mayor frecuencia disfunción de la médula ósea (principalmente leucopenia). En pacientes con mieloma múltiple, las reacciones adversas notificadas con mayor frecuencia (relacionadas con el tratamiento) en el tratamiento combinado con bortezomib fueron náuseas, diarrea, neutropenia, trombocitopenia, vómitos, fatiga y estreñimiento. Pacientes con cáncer de mama (la dosis de la preparación 50 mg / m2 cada 4 semanas). Muy frecuentes: anorexia, náuseas, estomatitis, vómitos, EPP, alopecia, erupción cutánea, astenia, fatiga, mucositis no especificada. Frecuentes: faringitis, leucopenia, anemia, neutropenia, trombocitopenia, parestesia, dolor abdominal, estreñimiento, diarrea, dispepsia, ulceración de la boca, piel seca, decoloración de la piel, cambios de pigmentación, eritema, erupción cutánea, debilidad, pirexia, dolor, foliculitis , infecciones por hongos, herpes de los labios (origen no herpético), infecciones del tracto respiratorio superior, neuropatía periférica, lagrimeo, visión borrosa, arritmia ventricular, epistaxis, dolor de boca, erupciones ampollosas, dermatitis, erupción eritematosa, enfermedad de las uñas, escamas piel, calambres en las piernas, dolor de huesos, dolor musculoesquelético, dolor de mamas, edema, hinchazón de las piernas. Poco frecuentes: somnolencia. Pacientes con cáncer de ovario (la dosis de la preparación 50 mg / m2 cada 4 semanas). Muy frecuentes: leucopenia, anemia, neutropenia, trombocitopenia, anorexia, estreñimiento, diarrea, náuseas, estomatitis, vómitos, eritrodisestesia palmo-plantar (síndrome mano-pie; EPP), alopecia, erupción cutánea, debilidad, trastornos de las mucosas. Frecuentes: faringitis, parestesia, somnolencia, dolor abdominal, dispepsia, ulceración de la boca, piel seca, decoloración de la piel, pirexia, dolor, infección, candidiasis oral, herpes, infección del tracto urinario, anemia hipocrómica, reacciones alérgicas, deshidratación, caquexia, ansiedad. , depresión, insomnio, dolor de cabeza, mareos, neuropatía, hipertonía, conjuntivitis, trastornos cardiovasculares, vasodilatación, disnea, aumento de la tos, ulceración de la boca, esofagitis, gastritis, disfagia, sequedad de boca, flatulencia, gingivitis, disgeusia, erupción vesicular, picazón, dermatitis exfoliativa, cambios en la piel, erupción maculopapular, sudoración, acné, ulceración de la piel, dolor de espalda, dolor muscular, dolor al orinar, vaginitis, escalofríos, dolor en el pecho, malestar, edema periférico, pérdida de peso. Pacientes con mieloma múltiple (dosis de 30 mg / m2 de la preparación en combinación con bortezomib en un ciclo de 3 semanas). Muy frecuentes: anemia, neutropenia, trombocitopenia, anorexia, neuropatía sensorial periférica, neuralgia, dolor de cabeza, náuseas, diarrea, vómitos, estreñimiento, estomatitis, EPP, erupción cutánea, astenia, fatiga, pirexia. Frecuentes: herpes, herpes zoster, leucopenia, disminución del apetito, insomnio, neuropatía periférica, neuropatía, parestesia, polineuropatía, mareos, disgeusia, disnea, dolor abdominal, dispepsia, piel seca, dolor en las extremidades, pérdida de peso, neumonía, nasofaringitis. , infección del tracto respiratorio superior, candidiasis oral, neutropenia febril, linfopenia, deshidratación, hipopotasemia, hiperpotasemia, hipomagnesemia, hiponatremia, hipocalcemia, ansiedad, letargo, hipoestesia, síncope, disestesia, conjuntivitis, hipotensión, hipotensión arterial, enrojecimiento de la piel, hipertensión, flebitis, tos, epistaxis, disnea de esfuerzo, dolor en el tracto gastrointestinal superior, ulceración de la boca, sequedad de boca, disfagia, estomatitis aftosa, picor, urticaria papular, dermatitis alérgica, eritema, hiperpigmentación de la piel, equimosis puntiformes, alopecia, erupción por medicamentos, artralgia, mialgia, espasmos musculares, debilidad muscular, dolor musculoesquelético, dolor musculoesquelético en el pecho, eritema escrotal, edema periférico, escalofríos, síntomas de paragripe, malestar general, hipertermia, aumento de los niveles de AST , disminución de la fracción de eyección del miocardio, aumento de la creatinina, aumento de la ALT. Pacientes con sarcoma de Kaposi en curso de sida (dosis de preparación 20 mg / m2 cada 2-3 semanas). Muy frecuentes: neutropenia, anemia, leucopenia, náuseas. Frecuentes: candidiasis oral, trombocitopenia, anorexia, mareos, retinitis, vasodilatación, disnea, diarrea, gastritis, vómitos, ulceración de la boca, dolor abdominal, glositis, estreñimiento, náuseas, vómitos, alopecia, erupción cutánea, debilidad, fiebre, reacciones agudas a la infusión, pérdida de peso. Poco frecuentes: confusión, alteraciones sensoriales, eritema de palmas y plantas (EPP). También se han observado reacciones de hipersensibilidad que incluyen reacciones anafilácticas (infecciones por Pneumocystis carinii, Mycobacterium avium) y se observan con frecuencia en pacientes con inmunodeficiencia inducida por VIH. Todos los grupos de pacientes. Reacciones relacionadas con la perfusión: reacciones de hipersensibilidad, reacciones anafilactoides, broncoespasmo, edema facial, hipotensión, vasodilatación, urticaria, dolor de espalda, dolor de pecho, escalofríos, pirexia, hipertensión, taquicardia, indigestión, náuseas, mareos, dificultad respiratoria, faringitis, erupción cutánea, picazón, sudoración, reacciones en el lugar de la inyección e interacciones medicamentosas. En muy raras ocasiones, se han notificado convulsiones asociadas con reacciones relacionadas con la perfusión. Todos los pacientes experimentaron reacciones relacionadas con la perfusión principalmente durante la primera perfusión. La interrupción temporal de la perfusión suele corregir estos síntomas sin necesidad de más tratamiento. En casi todos los pacientes, el tratamiento con la preparación se puede reanudar después de que los síntomas se hayan resuelto sin recurrencia. Las reacciones relacionadas con la perfusión rara vez ocurren con los ciclos de tratamiento posteriores. Se ha notificado disfunción de la médula ósea que conduce a anemia, trombocitopenia, leucopenia y, en raras ocasiones, neutropenia febril. Se ha notificado con frecuencia estomatitis en pacientes que reciben infusión continua. Se ha observado un aumento en la incidencia de ICC con el tratamiento con doxorrubicina a una dosis acumulada> 450 mg / m2. en la vida o en una dosis más baja en pacientes con riesgo de desarrollar complicaciones del músculo cardíaco. Se han observado leucemias mieloides agudas secundarias y mielodisplasia en pacientes que reciben terapia combinada con doxorrubicina. Muy raramente se han observado cambios necróticos locales resultantes de la extravasación (en caso de síntomas, la perfusión debe detenerse inmediatamente y el resto del fármaco administrado en otra vena). En raras ocasiones, se ha producido la reaparición de lesiones cutáneas debido a radioterapia previa. En la experiencia postcomercialización se han notificado muy raramente enfermedades cutáneas graves (eritema multiforme, síndrome de Stevens-Johnson y necrólisis epidérmica tóxica). Se han notificado casos raros de tromboembolismo venoso, que incluyen tromboflebitis, trombosis venosa y embolia pulmonar (debido a que los pacientes con cáncer tienen un mayor riesgo de tromboembolismo, no se puede establecer una relación causal con el uso de la preparación). Muy raramente se han observado cambios necróticos resultantes de la extravasación.
Embarazo y lactancia
No lo use durante el embarazo, a menos que sea claramente necesario (existe riesgo de defectos de nacimiento graves en el feto). Las mujeres en edad fértil deben evitar quedarse embarazadas mientras ellas o sus parejas reciben tratamiento y durante 6 meses después de suspender el tratamiento. Debe interrumpirse la lactancia antes de iniciar el tratamiento con la preparación. Las mujeres infectadas por el VIH no deben amamantar a sus hijos bajo ninguna circunstancia para evitar la transmisión de madre a hijo.
Comentarios
La influencia de la preparación sobre la capacidad para conducir vehículos y utilizar máquinas es nula o insignificante. Sin embargo, los pacientes deben evitar conducir u operar maquinaria si experimentan mareos o somnolencia.
Interacciones
Se debe tener precaución al coadministrar medicamentos que interactúan con el clorhidrato de doxorrubicina estándar. La preparación puede aumentar la toxicidad de otros tratamientos contra el cáncer. No se observó toxicidad adicional en pacientes con tumores sólidos (incluido cáncer de mama y ovario) tratados simultáneamente con ciclofosfamida o taxanos durante los ensayos clínicos. En pacientes con SIDA, se ha informado que el clorhidrato de doxorrubicina estándar potencia la cistitis hemorrágica inducida por ciclofosfamida y potencia la hepatotoxicidad de la 6-mercaptopurina. Se debe tener precaución al usar cualquier otro fármaco citotóxico al mismo tiempo, especialmente uno que sea perjudicial para la función de la médula ósea.
La preparación contiene la sustancia: clorhidrato de doxorrubicina.
Medicamento reembolsado: NO